

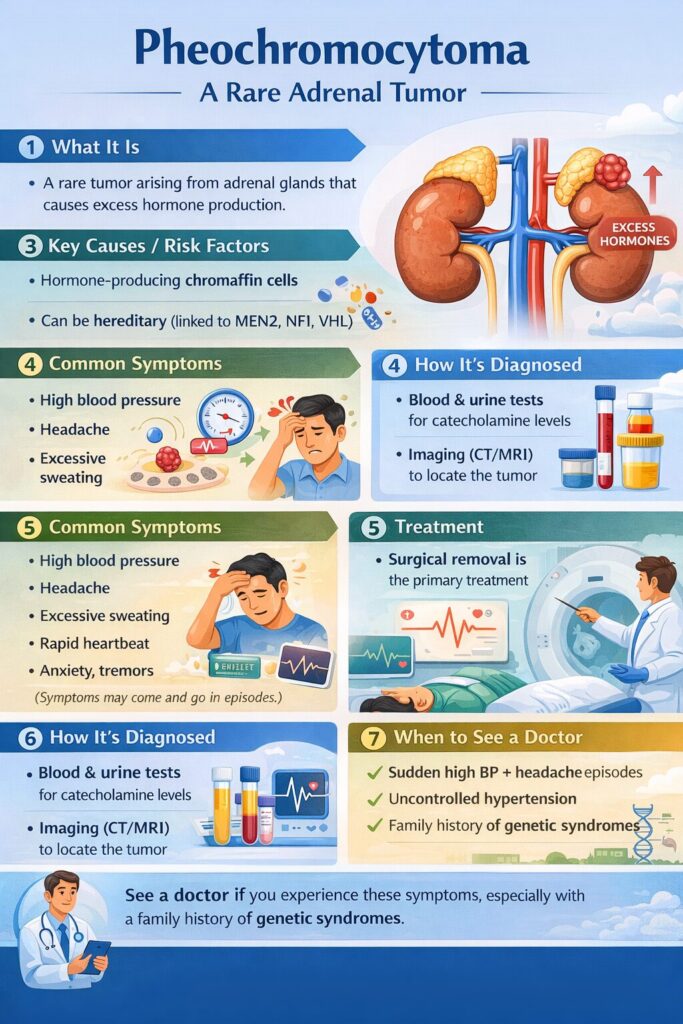
Pheochromocytoma is a rare tumor that develops in the adrenal glands and causes excessive production of hormones like adrenaline and noradrenaline. These hormones can lead to dangerously high blood pressure, heart complications, headaches, sweating, and anxiety attacks. Early diagnosis and proper treatment are essential to prevent life-threatening complications.
This guide explains what pheochromocytoma is, its causes, symptoms, diagnosis, treatment options, surgery cost in India, recovery, and frequently asked questions, helping patients and caregivers make informed medical decisions.
Pheochromocytoma, a rare yet potentially life-threatening tumor of the adrenal glands, poses a complex challenge for both patients and healthcare professionals. This enigmatic condition arises from chromaffin cells found in the adrenal medulla and results in the excessive release of adrenaline & noradrenaline, leading to a myriad of symptoms that can significantly impact an individual’s quality of life.
Help patients access appropriate treatment pathways when you Refer a Patient for Medical Help promptly.
In this comprehensive blog, we delve into the complexity of pheochromocytoma, exploring its symptoms, meaning, diagnostic approaches, and the diverse array of treatment options available.
Pheochromocytoma derives its name from the chromaffin cells (“pheo-” from the Greek word for dusky) that it originates from within the adrenal glands. These cancer tumors are typically benign but can be malignant in rare cases.
The adrenal medulla’s abnormal growth triggers an overproduction of catecholamines, such as adrenaline and noradrenaline, leading to a surge in blood pressure and a range of systemic effects.
Once diagnosed, the management of pheochromocytoma involves a multi-faceted approach. Surgical removal of the tumor, known as adrenalectomy, is the primary treatment, often resulting in a cure for the majority of patients.
However, the perioperative period poses unique challenges due to the risk of hypertensive crises during the manipulation of the tumor. Preoperative alpha-adrenergic blockade is commonly employed to mitigate these risks.
Pheochromocytoma remains a fascinating yet challenging clinical entity, requiring a proper understanding of its symptoms, diagnostic intricacies, and different treatment options.
By finding the complexities surrounding this adrenal gland tumor, healthcare professionals can better guide patients towards effective management strategies, ultimately improving outcomes and quality of life for those affected by this rare condition.
Through crowdfunding donation, financial goals become more achievable.
Community backing strengthens every effort.
Discover patient-centered care insights once you Explore Patient Care Guides thoroughly.

Table of Contents
- What Is Pheochromocytoma?
- Pheochromocytoma Symptoms
- Pheochromocytoma Meaning
- Pheochromocytoma Diagnosis
- Pheochromocytoma Treatment Cost in India
- Best Cities in India for Pheochromocytoma Treatment
- Why Is Surgery Required for Pheochromocytoma?
- Pheochromocytoma Treatment
- Pre-Surgery Preparation for Pheochromocytoma
- What To Expect After Pheochromocytoma Treatment?
- Recovery After Pheochromocytoma Surgery
- Neuroblastoma Vs Pheochromocytoma
- Pheochromocytoma Causes
- Pheochromocytoma Life Expectancy
- Complications Of Pheochromocytoma
- Risks and Complications
- Need Financial Help for Pheochromocytoma Treatment?
- Medical Review & Content Accuracy
- Conclusion
What Is Pheochromocytoma?
Pheochromocytoma is a rare, usually non-cancerous tumor of the adrenal glands that causes excess hormone release, leading to severe hypertension, headaches, palpitations, sweating, and anxiety. It is primarily treated through surgical removal of the tumor.
Pheochromocytoma Symptoms
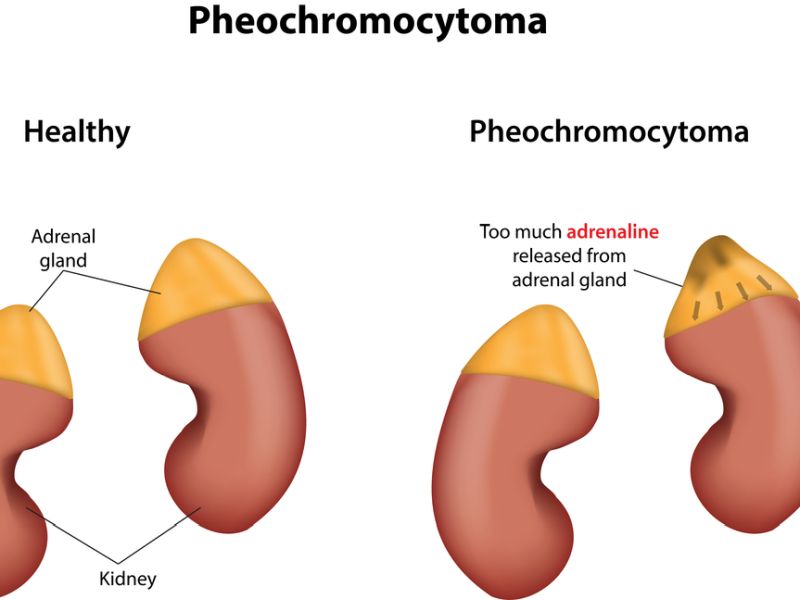
Pheochromocytoma symptoms is a rare cancer tumor that develops in the adrenal glands, which are situated on top of each kidney. These tumors are typically non-cancerous (benign) but can cause significant health issues due to the excessive production of catecholamines, like adrenaline & noradrenaline.
These hormones play a vital part in the body’s “fight or flight” response, regulating heart rate, blood pressure, and metabolism. When a pheochromocytoma releases excessive amounts of these hormones, it can lead to a combination of symptoms. Here’s a detailed explanation of the symptoms associated with pheochromocytoma:
1. Hypertension (High Blood Pressure): Pheochromocytomas often cause episodes of severe hypertension. The elevated levels of adrenaline and noradrenaline lead to increased heart rate and force of heart contractions, resulting in high blood pressure. The hypertension is usually episodic and can be paroxysmal, meaning it comes and goes.
2. Headaches: Persistent and severe headaches are common symptoms of pheochromocytoma. These headaches are often described as throbbing & may be accompanied by other neurological symptoms.
3. Sweating and Flushing: Excessive sweating, particularly in stressful situations or during episodes of high blood pressure, is a hallmark symptom. The skin may also become flushed or reddened during these episodes.
4. Palpitations and Rapid Heartbeat: Patients with pheochromocytoma may experience palpitations or a pounding sensation in the chest. The heart rate can become very rapid during episodes of elevated catecholamines.
5. Tremors and Shaking: Fine tremors or shaking of the hands and other parts of the body may occur. This is often related to the increased sympathetic nervous system activity.
6. Anxiety and Nervousness: Patients may feel a heightened sense of anxiety, nervousness, or a feeling of impending doom during episodes of catecholamine release.
7. Weight Loss: Unexplained weight loss may happen due to the growing metabolic rate associated with elevated levels of adrenaline and noradrenaline.
8. Abdominal Pain: Some patients may experience abdominal pain or discomfort, often in the region of the adrenal glands.
9. Chest Pain: Chest pain or discomfort may occur, especially during episodes of high blood pressure and increased cardiac activity.
10. Nausea and Vomiting: Gastrointestinal symptoms such as nausea and vomiting may occur, particularly during hypertensive crises.
It’s important to note that the symptoms of pheochromocytoma can be intermittent and may vary in severity. Additionally, the classic triad of symptoms includes headache, palpitations, and sweating. However, not all patients with pheochromocytoma will exhibit all these symptoms, and the presentation can be diverse.
If pheochromocytoma is suspected based on symptoms, diagnostic tests such as blood and urine catecholamine measurements, as well as imaging studies like CT (Computed Tomography) scans or MRI, are typically performed to confirm the diagnosis.
Fundraise online and manage your campaign with real-time tracking.
Monitoring progress becomes effortless.
Evaluate cost expectations clearly through the Disease Cost Calculator tool.
Pheochromocytoma Meaning
Pheochromocytoma is composed of chromaffin cells, which are specialized cells that make and release catecholamines. Catecholamines are hormones such as adrenaline (epinephrine) and noradrenaline (norepinephrine) that stimulate the sympathetic nervous system, which is accountable for the “fight or flight” response.
Heochromocytoma can occur at any age but is more common in adults between 20 and 50 years old. When a pheochromocytoma produces excess catecholamines, it causes symptoms like high blood pressure, rapid heartbeat, sweating, headache, and anxiety.
Pheochromocytoma can occur in one or both adrenal glands or in other parts of the body where chromaffin cells are present. When a pheochromocytoma occurs outside the adrenal glands, it is called a paraganglioma.
Paragangliomas can be found in the head and neck region, the chest, the abdomen, or the pelvis. Some paragangliomas may not produce catecholamines or may produce other substances, such as dopamine or serotonin.
Pheochromocytoma can be sporadic or familial. Sporadic means it occurs by chance and is not inherited. Familial means it runs in families and is caused by a genetic mutation.
About 10% to 25% of pheochromocytomas are familial and may be associated with other genetic syndromes like neurofibromatosis type 1 (NF1), multiple endocrine neoplasia type 2 (MEN2), von Hippel-Lindau disease (VHL), or hereditary paraganglioma syndromes (HPS).
Raise donations online by sharing clear goals and updates.
Transparency builds donor trust.
Simplify complex care decisions once you Find hospital and fund your treatment efficiently.
Pheochromocytoma Diagnosis
Diagnosing pheochromocytoma involves a combination of clinical evaluation, biochemical testing, and imaging studies. Here is a detailed explanation of the diagnostic process:
1. Clinical Evaluation:
– Medical History: A complete medical history is crucial, as the symptoms of pheochromocytoma can be nonspecific and overlap with other conditions. Symptoms may include high blood pressure, headaches, sweating, palpitations, and anxiety attacks.
– Family History: Pheochromocytoma can be related to certain genetic syndromes, like multiple endocrine neoplasia type 2 (MEN 2) and von Hippel-Lindau (VHL) syndrome. A family history of these syndromes may raise suspicion.
2. Biochemical Testing:
– Plasma or Urinary Metanephrines and Normetanephrines: These are breakdown products of catecholamines. Elevated levels of metanephrines and normetanephrines in blood or urine strongly suggest the presence of a pheochromocytoma. 24-hour urine collection or plasma-free metanephrines are commonly used tests.
– Catecholamines: Measurement of catecholamines (such as adrenaline and noradrenaline) in blood or urine can also aid in the diagnosis.
3. Imaging Studies:
– CT Scan or MRI: These imaging studies can help locate the tumor & determine its size. A contrast-enhanced Computed Tomography or MRI of the abdomen & pelvis is typically performed. Pheochromocytomas often appear as solid, well-defined masses.
– MIBG Scintigraphy: Metaiodobenzylguanidine (MIBG) is a radioactive substance that is taken up by certain tumors, including pheochromocytomas. MIBG scintigraphy involves injecting a small amount of radioactive MIBG and then performing a scan to identify tumor locations.
– PET Scan: Positron emission tomography (PET) scans using specific tracers can also help locate pheochromocytomas.
4. Functional Localization:
– Selective Venous Sampling: In some cases, especially when imaging results are inconclusive, selective venous sampling may be performed. This involves sampling blood from different veins to identify the site of excess catecholamine production.
– Octreotide Scintigraphy: This nuclear medicine imaging technique uses a radioactive substance called octreotide, which binds to certain tumors, including pheochromocytomas.
5. Genetic Testing:
– If there is a suspicion of an underlying genetic syndrome, genetic testing may be recommended to identify mutations associated with conditions like MEN 2 or VHL.
6. Provocative Testing:
– Some patients may undergo provocative testing, such as the clonidine suppression test or the glucagon stimulation test, to induce the release of catecholamines and confirm the diagnosis.
It’s important to note that the diagnostic process may vary for each individual, and the healthcare team will tailor the approach based on the patient’s specific symptoms and circumstances. Once diagnosed, prompt treatment, usually involving surgical removal of the tumor, is essential to manage the condition and prevent complications.
When you raise money, you’re opening the door to new possibilities.
Through crowdfunding, others step forward to help you succeed.
Pheochromocytoma Treatment Cost in India
The cost of treating pheochromocytoma in India depends on hospital location, surgical approach, surgeon expertise, ICU requirement, and complications.
| Treatment | Estimated Cost in India |
|---|---|
| Diagnosis & Imaging Tests | ₹15,000 – ₹40,000 |
| Pre-operative Medical Management | ₹20,000 – ₹60,000 |
| Laparoscopic Adrenalectomy | ₹1.8 – ₹3.5 lakh |
| Open Surgery (Complex Cases) | ₹2.5 – ₹5 lakh |
| ICU & Hospital Stay | ₹30,000 – ₹1 lakh |
Costs may vary based on hospital, city, patient condition, and complications.
Best Cities in India for Pheochromocytoma Treatment
- Mumbai: Advanced endocrine surgery centers, multispeciality hospitals
- Delhi NCR: High-volume endocrine surgery units, tertiary hospitals
- Chennai: Leading endocrine and oncology hospitals
- Bangalore: Robotic and minimally invasive adrenal surgery centers
- Hyderabad: High-success endocrine surgical programs
- Kochi: Advanced laparoscopic adrenalectomy centers
Why Is Surgery Required for Pheochromocytoma?
Surgical removal of the tumor is the definitive treatment for pheochromocytoma because medications alone cannot cure the condition. Surgery helps:
- Normalize blood pressure
- Prevent heart attack and stroke
- Eliminate hormone overproduction
- Reduce long-term complications
- Improve quality of life
Pheochromocytoma Treatment
The main pheochromocytoma treatment is surgery to remove the tumor. However, before surgery, the patient needs to take medications to control the blood pressure and prevent complications during the operation. The medications used are alpha-blockers and beta-blockers, which block the effects of adrenaline & noradrenaline on the blood vessels and the heart.
Surgery for pheochromocytoma can be performed using two methods:
– Laparoscopic surgery: This is a minimally invasive method that uses small incisions and a camera to guide the surgeon in removing the tumor. Laparoscopic surgery has different advantages over open surgery, such as less pain, faster recovery, and lower risk of infection or bleeding.
Open surgery: This is a traditional technique that uses a large incision to access and remove the tumor. It may be required if the tumor is large, malignant, or located in a difficult area.
Most pheochromocytomas are benign and do not recur after surgery. However, some tumors may be malignant or metastatic, meaning they have spread to other organs. In these cases, additional treatments may be needed, such as:
– MIBG therapy: This is a form of radiation therapy that combines MIBG, a compound that attaches to adrenal tumors, with a type of radioactive iodine. The radioactive iodine delivers radiation to the tumor cells and kills them.
– Peptide receptor radionuclide therapy (PRRT): This therapy combines a drug that targets cancer cells and a small amount of a radioactive substance. The radioactive substance delivers radiation to the tumor cells and kills them.
Chemotherapy: It involves the use of drugs that kill cancer tumors or stop them from growing. It may be given orally or intravenously.
Cost of Pheochromocytoma Treatment in India
The average cost of pheochromocytoma treatment in India ranges from INR 2 lakhs to INR 5 lakhs (USD 2700 to USD 6800). The cost of pheochromocytoma treatment in India depends on various factors, such as:
– The type and stage of the tumor
– The choice of hospital and surgeon
– The type of surgery and anesthesia
– The need for additional treatments or tests
– The duration of hospital stay and recovery
The approximate cost of pheochromocytoma treatment in India is as follows:
– Laparoscopic surgery: Rs. 1,50,000 to Rs. 2,50,000
– Open surgery: Rs. 2,00,000 to Rs. 3,00,000
– MIBG therapy: Rs. 3,00,000 to Rs. 5,00,000
– PRRT therapy: Rs. 5,00,000 to Rs. 10,00,000
– Chemotherapy: Rs. 50,000 to Rs. 1,00,000 per cycle
These costs are only indicative and may vary depending on the individual case and the hospital policy.
Pre-Surgery Preparation for Pheochromocytoma
Before surgery, patients require 10–14 days of medical preparation to stabilize blood pressure and heart rate.
This includes:
- Alpha-blockers
- Beta-blockers (if needed)
- High-salt diet and hydration
- Continuous BP and heart monitoring
Proper preparation significantly reduces surgical risk.
What To Expect After Pheochromocytoma Treatment?
After pheochromocytoma treatment, here are some general expectations:
1. Recovery from Surgery:
– The most common treatment for pheochromocytoma is surgical removal of the tumor. After surgery, there is a recovery period, and the duration can differ depending on the individual and the extent of the surgery.
– You may experience some pain and discomfort at the surgical site, & your medical team will give pain management guidelines for recovery.
2. Blood Pressure Management:
– Pheochromocytomas can cause high blood pressure due to the excess production of adrenaline. After the tumor is removed, blood pressure often returns to normal. However, some individuals may still experience blood pressure fluctuations, and ongoing monitoring is essential.
– Medications may be recommended to manage blood pressure, especially during the postoperative period. It’s important to take these medications as directed.
3. Hormone Level Stabilization:
– Removing the pheochromocytoma eliminates the source of excess adrenaline production. Hormone levels should gradually stabilize, leading to a reduction in symptoms such as anxiety, palpitations, and sweating.
4. Follow-up Monitoring:
– Periodic follow-up appointments with your medical team are necessary for monitoring blood pressure, hormone levels, and overall health.
– Follow-up imaging studies may be conducted to ensure that there is no recurrence of the tumor.
5. Lifestyle Changes:
– Following a healthy lifestyle is crucial for managing overall well-being. This includes maintaining a balanced diet, daily exercise, & managing stress.
– It’s important to avoid triggers that can elevate blood pressure, such as certain medications or dietary factors.
6. Potential Long-Term Effects:
– While the removal of the pheochromocytoma typically results in a good prognosis, there may be long-term effects. Some patients may persist in experiencing residual symptoms or have ongoing challenges with blood pressure management.
7. Psychological Support:
– Dealing with a rare condition like pheochromocytoma and its treatment can be emotionally challenging. Psychological support, either through counseling or support groups, may be beneficial for coping with the experience.
It’s vital to note that individual experiences can differ, and your healthcare team will provide personalized guidance based on your specific case. Regular communication with your healthcare providers, adherence to recommended follow-up appointments, and compliance with any prescribed medications are crucial for ongoing health management.
Recovery After Pheochromocytoma Surgery
- Hospital stay: 3–7 days
- Return to daily activities: 2–3 weeks
- Complete recovery: 4–6 weeks
- Blood pressure normalization: Usually within days
Regular follow-ups and hormone monitoring are required to prevent recurrence.
Neuroblastoma Vs Pheochromocytoma
Neuroblastoma and pheochromocytoma are both tumors that arise from neural crest cells, but they differ in various aspects, including their locations, characteristics, and clinical presentations.
1. Origin and Cell Type:
– Neuroblastoma: It originates from embryonic nerve tissue, particularly the neural crest cells that give elevation to the sympathetic nervous system. Neuroblastomas often occur in the adrenal glands but can also develop along the sympathetic chain in the abdomen, chest, pelvis, or neck.
Pheochromocytoma: This tumor arises from chromaffin cells in the adrenal medulla, the major part of the adrenal glands. These cells produce & release hormones such as adrenaline and noradrenaline.
2. Age of Onset:
– Neuroblastoma: It is most commonly found in infants and young children, usually before the age of 5. However, it can occasionally occur in older children and adults.
– Pheochromocytoma: It typically presents in adults, with the majority of cases diagnosed between the ages of 30 and 50. However it can also occur in children, but it is relatively rare in this age group.
3. Clinical Presentation:
– Neuroblastoma: Symptoms may vary widely but can include abdominal distension, pain, changes in bowel habits, and neurological symptoms due to the tumor’s location and potential spread to other areas of the body.
– Pheochromocytoma: The classic triad of symptoms includes headaches, palpitations, and sweating. Patients may also experience high blood pressure, anxiety, and weight loss.
4. Hormone Production:
– Neuroblastoma: While some neuroblastomas can produce hormones, it is not a consistent feature. Hormone production, when present, may lead to symptoms such as diarrhea, flushing, or high blood pressure.
– Pheochromocytoma: These tumors almost always produce excessive amounts of catecholamines (adrenaline and noradrenaline), leading to the characteristic symptoms mentioned above.
5. Malignancy and Prognosis:
– Neuroblastoma: It can vary widely in terms of aggressiveness. Some neuroblastomas are benign or have a favorable prognosis, while others can be highly malignant and aggressive.
– Pheochromocytoma: The majority of pheochromocytomas are benign, but they can be locally invasive. Malignant cases are rare but have a poorer prognosis.
In summary, while both neuroblastoma and pheochromocytoma originate from neural crest cells, they have distinct clinical features, age distributions, and prognoses. Neuroblastoma is more common in children and can occur in various locations, while pheochromocytoma typically presents in adults and arises from the adrenal medulla, producing excess catecholamines.
Pheochromocytoma Causes
The exact cause of pheochromocytoma is not always clear, but there are some factors and conditions associated with its development:
1. Genetic Factors: In some cases, pheochromocytoma may have a genetic component. Certain genetic syndromes are linked to an increased risk of developing these tumors. Examples include multiple endocrine neoplasia type 2 (MEN2), von Hippel-Lindau (VHL) syndrome, & neurofibromatosis type 1 (NF1).
2. Sporadic Cases: Pheochromocytomas can also occur sporadically without any known genetic predisposition. The reason for their development in these cases needs to be better understood.
3. Age and Gender: Pheochromocytomas can develop at any age, but they are most commonly diagnosed in people between the ages of 30 and 50. They affect men and women equally.
4. Other Endocrine Disorders: Certain hormonal disorders or imbalances may contribute to the development of pheochromocytoma. Conditions such as paraganglioma (a tumor that arises in the nerve cells) can also cause the overproduction of catecholamines.
5. Trauma: While rare, trauma or injury to the adrenal glands may trigger the development of pheochromocytoma in some cases.
6. Environmental Factors: There is no strong evidence linking environmental factors to the development of pheochromocytoma. However, exposure to certain toxins or chemicals may play a role in some cases.
Pheochromocytoma Life Expectancy
The life expectancy of people with pheochromocytoma depends on several factors, such as:
– The size and location of the tumor
– The presence or absence of metastasis (spread) to other organs
– The presence or absence of genetic syndromes
– The response to treatment
According to estimates, around 95% of people diagnosed with a benign pheochromocytoma that has not spread to other parts of their body live at least 5 more years. If the tumor is malignant and has spread or come back after treatment, nearly 34% to 60% of people live at least five years after diagnosis.
People who have a genetic syndrome that predisposes them to pheochromocytoma may have a higher risk of recurrence or developing other tumors in different organs. Therefore, they need regular follow-up and screening for life.
Early diagnosis & treatment can improve the prognosis of people with pheochromocytoma. Consequently, it is important to seek medical attention if you have any symptoms or risk factors for this condition.
Complications Of Pheochromocytoma
Some of the possible complications are:
– Acute hypertensive crisis: a sudden and severe increase in blood pressure that can harm the organs and tissues in the body
– Cardiovascular disease: damage to the heart & blood vessels due to chronic high blood pressure or acute hypertensive crisis
– Stroke: interruption of blood discharge to the brain due to a clot or bleeding
– Kidney failure: loss of kidney function caused by damage from high blood pressure or acute hypertensive crisis
– Acute respiratory distress: difficulty breathing due to fluid accumulation in the lungs
– Damage to the nerves of the eye: loss of vision or blindness due to high blood pressure or acute hypertensive crisis
To prevent these complications, it is vital to take medical attention if you have symptoms suggestive of pheochromocytoma, especially if you have a family history of the condition or a related genetic syndrome.

Risks and Complications
When performed by experienced surgeons, pheochromocytoma surgery is generally safe. Possible complications include:
- Sudden blood pressure fluctuations
- Bleeding
- Infection
- Hormonal imbalance
- Temporary low blood pressure
Proper pre-operative preparation and ICU monitoring significantly reduce these risks.
Need Financial Help for Pheochromocytoma Treatment?
Pheochromocytoma surgery and long-term treatment can be expensive and may cause financial strain.
ImpactGuru helps patients raise funds for rare disease treatments and complex surgeries through medical crowdfunding, enabling fast access to financial support with complete transparency.
👉 Start a Free Fundraiser for Treatment
Medical Review & Content Accuracy
This article is reviewed using verified endocrinology guidelines, endocrine surgery protocols, and publicly available clinical references to ensure accuracy, patient safety, and medical reliability. The content is intended for educational purposes only and does not replace professional medical consultation.
Pheochromocytoma is a rare tumour that develops in the adrenal glands and leads to the excessive release of hormones like adrenaline and noradrenaline, which can trigger pheochromocytoma symptoms such as high blood pressure, headaches, sweating, rapid heartbeat, and anxiety-like episodes.
This condition often arises due to pheochromocytoma causes linked to chromaffin cells and, in some cases, hereditary syndromes, and requires thorough pheochromocytoma diagnosis through blood or urine catecholamine measurements and imaging scans.
Managing the condition typically involves surgical removal of the tumour as part of pheochromocytoma treatment, supported by medications to control blood pressure beforehand. If left untreated, pheochromocytoma complications such as heart disease, stroke, and organ damage can occur, making early detection and intervention crucial for better outcomes.
Conclusion
Pheochromocytoma is a rare tumor in the adrenal glands that causes too much adrenaline production, leading to symptoms like high blood pressure, headaches, and sweating. The tumor’s main significance is that it interferes with the body’s adrenaline balance, causing specific symptoms. It usually appears in the adrenal glands but can sometimes appear elsewhere.
In summary, understanding pheochromocytoma’s symptoms, using the right tests, and choosing appropriate treatments are crucial for effective care. Early detection and proper management make a big difference for people dealing with this uncommon adrenal tumor.
Pheochromocytoma treatment often involves surgeries, medications, and follow-up care, all of which can result in substantial expenses. Many individuals and families may find it challenging to bear these costs alone, leading to financial strain and potential delays in seeking necessary medical care.
Crowdfunding harnesses the power of community, allowing friends, family, and even strangers to come together and support a person in need. Crowdfunding platforms have a global reach, enabling individuals to receive support from people worldwide.
FAQs
Most pheochromocytomas are benign (non-cancerous), but a small percentage can be malignant.
Yes, surgical removal usually provides a complete cure.
Recurrence is rare but possible, requiring long-term monitoring.
If untreated, it can cause severe hypertension, heart attack, stroke, and organ damage.
Endocrinologists and endocrine surgeons treat pheochromocytoma.










