

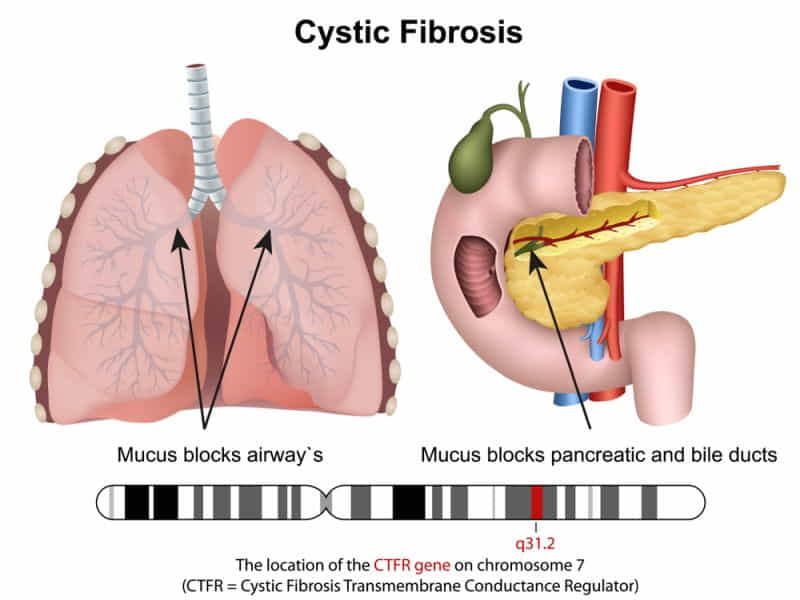
Cystic fibrosis is a genetic disorder that profoundly affects the respiratory, digestive, and reproductive systems. This condition arises from mutations in the CFTR gene, which is accountable for creating a protein crucial for maintaining the balance of salt & water on the surface of cells. As a result of these genetic anomalies, thick and sticky mucus accumulates in various organs, particularly the lungs and digestive tract, leading to many complications and health challenges.
The most common symptoms of cystic fibrosis manifest in the respiratory system, with chronic cough, recurrent lung infections, and difficulty breathing being prominent indicators. The thick mucus obstructs the airways, creating an ideal environment for bacterial growth, and as a consequence, individuals with CF often experience persistent respiratory infections. Over time, these recurrent infections contribute to progressive lung damage, significantly impacting respiratory function.
Beyond respiratory issues, cystic fibrosis also affects the digestive system. The thickened mucus obstructs the pancreatic ducts, preventing digestive enzymes from reaching the small intestine. This impedes the proper digestion and absorption of nutrients, leading to malnutrition, poor growth, and vitamin deficiencies. Additionally, CF can cause liver disease and gallbladder problems, further complicating the digestive challenges associated with the condition.
In the reproductive system, both male and female individuals with cystic fibrosis may encounter fertility issues. Men with CF often have a congenital absence of the vas deferens, which obstructs the passage of sperm. At the same time, women may face challenges due to thickened cervical mucus hindering sperm mobility.
Managing cystic fibrosis requires a comprehensive approach involving various therapeutic strategies. Physiotherapy and respiratory exercises are integral components to help clear mucus from the airways & improve lung function. Medications like bronchodilators and antibiotics are commonly prescribed to manage respiratory symptoms and prevent infections. Moreover, individuals with CF must adhere to a specialised diet with pancreatic enzyme supplements to aid digestion and ensure proper nutrient absorption.
The landscape of cystic fibrosis care has seen remarkable advancements in recent years. Innovative therapies, including CFTR modulator drugs, target the underlying genetic defects, providing unprecedented opportunities to address the root cause of the disease. These groundbreaking treatments aim to improve the quality of life for individuals with cystic fibrosis and offer hope for better outcomes in the long term.
Table of Contents
Cystic Fibrosis Symptoms
Cystic Fibrosis is a genetic disease primarily affecting the respiratory, digestive, & reproductive systems. It is led by mutations in the CFTR (cystic fibrosis transmembrane conductance regulator) gene. The symptoms of cystic fibrosis can differ widely, and their severity may also differ from person to person. Common symptoms include:
1. Respiratory Symptoms:
– Persistent coughing
– Wheezing
– Shortness of breath
– Recurrent lung infections (like pneumonia or bronchitis)
– Increased production of thick, sticky mucus
– Difficulty clearing mucus from the airways
2. Digestive Symptoms:
– Poor growth & weight gain despite a proper appetite
– Difficulty gaining and maintaining weight
– Frequent greasy, bulky stools
– Intestinal blockage (obstruction) due to thickened mucus
– Pancreatic insufficiency, leading to malabsorption of nutrients
3. Other Symptoms:
– Salty-tasting skin (individuals with CF may have higher than normal salt levels in their sweat)
– Clubbing of fingers and toes (enlargement of the fingertips and nails)
– Male infertility due to absence or blockage of the vas deferens
4. Complications:
– Diabetes: People with CF have an increased risk of developing diabetes due to pancreatic dysfunction.
– Liver disease: Some individuals may experience liver complications, including liver cirrhosis.
– Sinusitis: Chronic sinus infections can be a recurring issue.
If you suspect someone may have cystic fibrosis or if you are experiencing symptoms yourself, it is better to contact a medical professional for proper evaluation and diagnosis.
Cystic Fibrosis Treatments
The type of mutation a person inherits from their parents determines the severity of their symptoms and the treatment options that may be available. There is no cure for CF, but various treatments can help ease symptoms, reduce complications, and improve quality of life.
Medications
Medications are an essential part of CF treatment. They can target different aspects of the disease, such as:
– Medications targeting gene mutations: These drugs aim to rectify or improve the function of the defective CFTR protein. For example, Trikafta is a combination of three medicines that treats the most common genetic mutation causing CF, which affects about 90% of people with CF. Trikafta helps increase the amount and activity of the CFTR protein at the cell surface, allowing more chloride and water to move across the membrane. This reduces the thickness and stickiness of the mucus and improves lung function.
– Antibiotics: These drugs fight bacterial infections in the lungs, which are common and severe complications of CF. Antibiotics can be taken orally, inhaled, or injected into a vein. They can be used to treat acute infections or to prevent chronic illnesses. Some examples of antibiotics used for CF are azithromycin, ciprofloxacin, tobramycin, and ceftazidime.
– Mucolytics: These drugs thin the mucus and make it easier to cough up. They can be taken orally or inhaled. They can help clear the airways and prevent lung damage. Some examples of mucolytics used for CF are dornase alfa, hypertonic saline, and mannitol.
– Bronchodilators: These drugs relax and widen the muscles around the airways. They can be taken orally or inhaled. They can help improve airflow and reduce shortness of breath. Some examples of bronchodilators used for CF are albuterol, salmeterol, and ipratropium.
– Anti-inflammatories: These drugs reduce lung inflammation, which can cause swelling, pain, and tissue damage. They can be taken orally or inhaled. They can help prevent lung deterioration and improve lung function. Some examples of anti-inflammatories used for CF are ibuprofen, prednisone, and montelukast.
– Pancreatic enzymes: These supplements contain digestive enzymes that help break down food in the intestines. They can be taken orally with every meal and snack. They can help prevent malabsorption, malnutrition, and bowel problems. Some examples of pancreatic enzymes used for CF are Creon, Pancreaze, and Zenpep.
– Vitamins: These are supplements that contain vitamins that are essential for average growth and development. They can be taken orally once a day. They can help prevent vitamin deficiencies that can cause problems such as anaemia, osteoporosis, and bleeding disorders. Some examples of vitamins used for CF are ADEKs (vitamins A, D, E, and K), calcium, iron, and zinc.
Physiotherapy
Physiotherapy is another essential part of CF treatment. It involves various exercises and techniques that help clear the mucus from the lungs and improve breathing. Physiotherapy can be done at home or in a clinic with the help of a physiotherapist or a caregiver.
Some examples of physiotherapy methods for CF are:
– Chest physiotherapy (CPT): This technique involves tapping or vibrating the chest and back with a cupped hand or a device to loosen the mucus in the lungs. It can be done manually or with a mechanical device such as a vest or a flutter valve.
– Breathing exercises: These are exercises that involve inhaling deeply through the nose & exhaling forcefully through the mouth to move the mucus out of the lungs. They can be done with or without a device such as a PEP (positive expiratory pressure) mask or an acapella.
– Airway clearance devices: These help clear the mucus from the lungs by creating vibrations, oscillations, or pressure changes in the airways. They can be used alone or in combination with other methods. Some examples of airway clearance devices for CF are the Aerobika, the Lung Flute, and the Quake.
– Exercise: Any physical activity increases the heart and breathing rates. It can help improve lung function, muscle strength, endurance, and mood. It can also help prevent infections, diabetes, and osteoporosis. Some exercise examples for CF are walking, jogging, cycling, swimming, and dancing.
Lung Transplant
It is a medical procedure that involves replacing one or both diseased lungs from a donor with healthy lungs. It is considered a last resort treatment for people with CF who have severe lung damage that does not respond to other treatments.
A lung transplant can improve the quality and length of life for some people with CF, but it also carries many risks and challenges. Some of these are:
– Rejection: This is when the immune system attacks the transplanted lungs as foreign invaders. It can cause inflammation, scarring, and failure of the new lungs. It can be prevented or treated with immunosuppressive drugs, but these drugs can also cause side effects such as infections, diabetes, and cancer.
– Infection: This is when bacteria, viruses, fungi, or parasites invade the transplanted lungs or other body parts. It can cause fever, cough, chest pain, and breathing problems. It can be prevented or treated with antibiotics, antivirals, antifungals, or antiparasitics, but these drugs can also cause side effects like nausea, diarrhoea, and allergic reactions.
– Complications: These are problems that occur during or after the surgery. They can include bleeding, blood clots, stroke, heart attack, kidney failure, nerve damage, and death. They can be prevented or treated with blood transfusions, anticoagulants, painkillers, and supportive care.
Cystic Fibrosis (CF) Meaning
CF is a rare genetic disease that mainly affects the lungs & the pancreas, liver, kidneys, and intestine. The hallmark feature of CF is the accumulation of thick mucus in different organs. Long-term issues include difficulty breathing & coughing up mucus due to frequent lung infections.
CF is inherited from both parents, who usually carry a faulty gene that causes CF. Carriers do not have any symptoms of CF, but they can transfer the gene to their children. If both parents are carriers, there is a 25% probability that their child will have CF.
The gene that causes CF affects the production of a protein referred to as cystic fibrosis transmembrane conductance regulator (CFTR). This protein helps regulate salt and water movement across cell membranes. When CFTR is defective, the cells produce thick, sticky mucus clogging the tubes and ducts in different organs.
The most common organ affected by CF is the lungs. The mucus in the lungs makes it hard to breathe and creates a favourable environment for bacteria to grow. People with CF often have chronic lung infections, such as pneumonia or bronchitis, that damage the lung tissue over time. They also have frequent episodes of wheezing, shortness of breath, and coughing up blood.
Another organ affected by CF is the pancreas. The pancreas creates digestive enzymes that help to break down food in the small intestine. The mucus in the pancreas blocks the ducts that carry these enzymes to the intestine. As a result, people with CF have problems with digesting food and absorbing nutrients. They may experience symptoms such as abdominal pain, bloating, gas, diarrhoea, foul-smelling stools, poor weight gain, and growth failure.
Other organs that can be affected by CF include the liver, which can develop scarring and cirrhosis; the kidneys, which can build stones and infections; the intestines, which can create blockages and inflammation; the sinuses, which can develop polyps and illnesses; and the sex organs, which can cause infertility in both males and females.
Cystic Fibrosis Causes
Here are the key points regarding the causes of cystic fibrosis:
1. Genetic Inheritance:
– CF is an autosomal recessive genetic disease, meaning a person must inherit dual mutated copies of the CFTR gene (one from both parents) to develop the condition.
– Individuals who inherit one regular copy of the gene and one mutated copy are carriers but do not exhibit symptoms of cystic fibrosis.
2. CFTR Gene Mutations:
– The CFTR gene mutations produce a defective or malfunctioning CFTR protein.
– The normal CFTR protein plays a crucial role in controlling the movement of chloride ions across cell membranes. Dysfunction of this protein results in thick, sticky mucus accumulation in various organs.
3. Impact on Organ Function:
– The defective CFTR protein affects the function of cells in the respiratory, digestive, and reproductive systems.
– The abnormal mucus clogs airways in the lungs, leading to respiratory symptoms such as chronic coughing, recurrent lung infections, and difficulty breathing.
– In the digestive system, the thickened mucus can obstruct the pancreatic ducts, impairing the release of digestive enzymes & leading to malabsorption of nutrients.
– Other organs, such as the liver and reproductive organs, can also be affected.
4. Variability in Mutations:
– There are over 1,700 known mutations in the CFTR gene, & the severity of cystic fibrosis symptoms can vary depending on an individual’s specific mutation(s).
– Some mutations result in milder kinds of the disease, while others lead to more severe complications.
5. Newborn Screening:
– Cystic fibrosis can be diagnosed through newborn screening, which involves testing for elevated levels of immunoreactive trypsinogen (IRT) in a blood sample from a newborn. Subsequent genetic testing is then conducted to confirm the diagnosis.
Cystic Fibrosis Diagnosis
The diagnosis of CF can be challenging, as the symptoms and severity vary from person to person. However, various tests can help confirm the diagnosis and guide the treatment. Here are some of the main tests used to diagnose CF:
– Newborn screening: All newborns in the United States are tested for CF as part of routine screening. The screening involves a blood test that measures the amount of a chemical called immunoreactive trypsinogen (IRT) produced by the pancreas. High levels of IRT may indicate CF or other conditions that affect the pancreas. If the IRT test is positive, a second test is done to check for common CFTR mutations in the baby’s DNA. If both tests are positive, the diagnosis of CF is confirmed. Further testing is needed if only one test is positive or the results are unclear.
– Sweat test: This is the gold standard test for diagnosing CF. It measures the amount of salt (sodium chloride) in the sweat. People with CF have higher than normal salt levels in their sweat because their faulty CFTR protein does not allow enough water to pass through the cell membranes. The sweat test involves applying a chemical that stimulates sweating on a small skin area, usually on the arm or leg. Then, the sweat is collected on a special paper or pad and analysed for salt content. A sweat chloride level of over 60 millimoles per litre (mmol/L) is considered diagnostic of CF.
– Genetic testing: This test looks for specific mutations in the CFTR gene that cause CF. It can be done on a blood sample, saliva sample, or cells inside the cheek. Genetic testing can identify more than 2,000 different mutations but cannot detect all possible variants. Therefore, a negative genetic test does not rule out CF completely. Genetic testing can also decide if someone is a carrier of a CFTR mutation, meaning they have one normal copy & one faulty copy of the gene. Carriers do not have symptoms of CF, but they can pass on the mutation to their children.
Cystic Fibrosis Life Expectancy
Life expectancy depends on many factors, such as the type of mutation, the severity of symptoms, the availability and adherence to treatments, and other health conditions.
However, some statistics can give a general idea of how long people with CF can expect to live.As per the Cystic Fibrosis Foundation Patient Registry, the median anticipated survival age for patients with CF in the US based on their year of birth is:
– 31 years for those born between 1993 and 1997
– 37 years for those born between 2003 and 2007
– 44 years for those born between 2013 and 2017
The data also shows that half of all babies born with CF in 2017 will live to be 46 or older. These numbers have increased dramatically over the past few decades thanks to advances in screening and treatments. The latest research suggests that by 2025, the no. of adults living with CF will increase by around 75%.
These statistics reflect present life expectancy rates in developed countries like the US and the UK. However, in developing countries like El Salvador & India, life expectancy for people with CF may be under 15 years of age.
It is important to note that these figures are just averages. Some people will live longer. Some people with CF are living into their 70s. It is also important to note that these predictions do not consider the potential for improvements in care & treatment that may occur as people age.
Complications Of Cystic Fibrosis
The complications associated with cystic fibrosis can vary in severity and may affect multiple organ systems. Here are some common complications:
1. Respiratory Complications:
– Chronic Respiratory Infections: Thick mucus in the airways offers an ideal environment for the development of bacteria, leading to frequent lung infections.
– Bronchiectasis: Chronic inflammation and recurrent infections can cause damage to the airways, leading to the widening and scarring of the bronchi.
– Respiratory Failure: Over time, progressive lung damage may result in respiratory failure, requiring interventions such as oxygen therapy or lung transplantation.
2. Digestive Complications:
– Pancreatic Insufficiency: The pancreas may not produce enough enzymes to digest food properly, leading to malabsorption of nutrients and malnutrition.
– Liver Disease: CF can cause liver problems, including cirrhosis and an increased risk of liver infections.
3. Nutritional Complications:
– Malnutrition: Difficulty absorbing nutrients can lead to malnutrition, growth failure, and weight loss.
– Failure to Thrive: Infants and children with CF may experience failure to thrive due to nutritional challenges.
4. Reproductive Complications:
– Fertility Issues: Both men and women with CF may experience fertility problems, including reduced fertility and an increased risk of infertility.
5. Other Complications:
– Sinusitis: Thick mucus can also affect the sinuses, leading to chronic sinus infections.
– Bone Health: People with CF may have issues with bone health, including a higher risk of osteoporosis.
6. Psychosocial Complications:
– Emotional and Psychological Impact: Managing a chronic condition like CF can take a toll on mental health, leading to stress, anxiety, and depression.
7. Diabetes: Some individuals with CF may develop cystic fibrosis-related diabetes (CFRD) due to pancreatic insufficiency and insulin resistance.
8. Complications Related to Treatments:
– Antibiotic Resistance: Frequent use of antibiotics to manage respiratory infections may contribute to the development of antibiotic-resistant strains of bacteria.
– Side Effects of Medications: Some medications used to manage CF symptoms may have side effects.
Conclusion
In conclusion, cystic fibrosis is a complex genetic disorder that significantly impacts the respiratory and digestive systems. The disease arises from mutations in the CFTR gene, leading to the production of thick and sticky mucus in various organs. While there is no cure for cystic fibrosis, advancements in medical research have resulted in effective treatments and therapies that aim to manage symptoms, improve quality of life, and extend lifespan. As we continue to gain insights into the genetic basis of the disease, ongoing research offers hope for the development of innovative therapies and personalised treatments, bringing us closer to improved outcomes for those affected by cystic fibrosis.
Cystic Fibrosis not only poses a health challenge but also places a significant strain on financial resources. A fundraising website fills this gap by offering a platform where individuals can raise funds and seek financial support from a caring community, irrespective of their background or financial standing. Crowdfunding makes expensive healthcare more accessible.










